GEOmetadb is an attempt to make access to the metadata associated with the NCBI Gene Expression Omnibus (GEO) samples, platforms, and datasets much more feasible for common biologists and bioinformatians/statistians.
BEST (Bayesian Expression Search Tool) implements a model-based Bayesian querying tool. This program is designed to query large and heterogeneous microarray gene expression database. It is able to identify a small set of genes that share correlated gene expression profiles with the query gene under a subset of samples/experimental conditions. In addition, it allows linearly transformed expression patterns to be recognized and is robust against sporadic outliers in the data.
Bang is a fast repeat-supressing search tool, written primarily for anchoring reads to genomes but also adaptable to other genome scale comparison problems.
CLAST (CUDA implemented large-scale alignment search tool) is a tool that enables analyses of millions of reads and thousands of reference genome sequences, and runs on NVIDIA Fermi architecture graphics processing units.
GHOST-MP is a parallel sequence similarity search tool. It searches for similar sequences among nucleotide query sequences and amino acid sequence database like BLASTX.
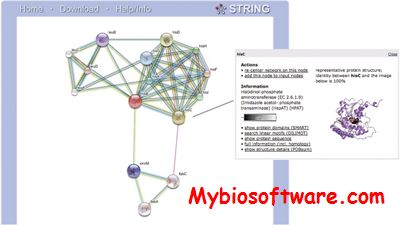
STRING (search tool for recurring instances of neighbouring genes) is a database and web resource dedicated to protein-protein interactions, including both physical and functional interactions. It weights and integrates information from numerous sources, including experimental repositories, computational prediction methods and public text collections, thus acting as a meta-database that maps all interaction evidence onto a common set of genomes and proteins.
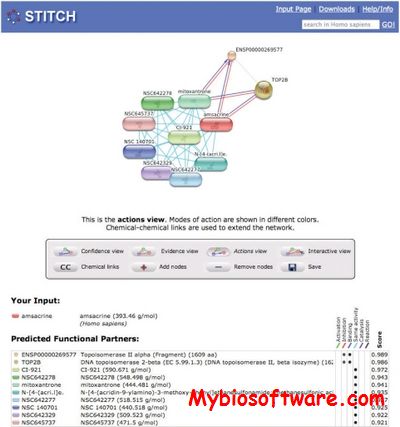
STITCH (search tool for interactions of chemicals) is a resource to explore known and predicted interactions of chemicals and proteins.Chemicals are linked to other chemicals and proteins by evidence derived from experiments, databases and the literature.
Peppy: a fully integrated, proteogenomic mapping software tool. Peppy generates a peptide database from genome files, tracks peptide loci, matches peptides to MS/MS spectra, assigns scores to those matches, and tracks score distributions to find E-Values. To determine false discovery rate thresholds, Peppy automatically generates a decoy database from the original genome files and repeats the entire search process.
The FingerPRINTScan package uses an approach, based on Karlin and Altschul statistics, to generate a probability-value for each scoring match to a motif.