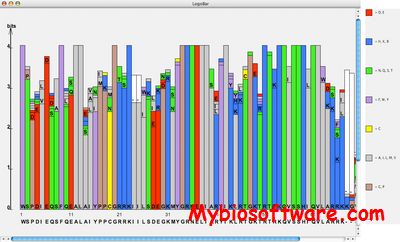
LogoBar is a Java application to display protein sequence logos. With this application you can generate protein sequence logos from multiple sequence alignments on your own computer, and you will get an additional output file that shows the amino acid incidence at every position of the multiple sequence alignment.
Asymmetry programs: AmbiguityRemover removes ambiguously aligned sites from protein sequence alignments, for input into AsymmetryCounter. AsymmetryCounter counts the number of amino acid subsitutions in each direction between pairs of protein sequences. AsymmetryScaler takes the results from AsymmetryCounter and summarizes substitutional asymmetry data in a single scale
SEQSEE (SEQuence SEEr) is a multi-purpose menu-driven suite of programs designed to provide a fully integrated analysis of protein sequences and protein databases. It contains rapid database searching, flexible pattern matching and multiple sequence alignment as well as a large number of structural analysis and prediction programs.
ANTHEPROT (ANalyse THE PROTeins) is intended to perform protein sequence analysis with a high integration level and clients/server capabilities. It is an interactive program with a graphical user interface that allows handling of protein sequence and data in a very interactive and convenient manner. It provides many methods and tools, which are integrated into a graphical user interface.
The Paircoil2 program predicts coiled-coil domains in protein sequences by using pairwise residue correlations obtained from a coiled-coil database. The original Paircoil program is still available for use.
K-Pax contains an implementation of a Bayesian model-based method for simultaneously classifying aligned proteins into functionally divergent subgroups and identifying their function specific residues
DLocalMotif is a discriminitive motif discovery web service specifically designed to discover local motifs in protein sequences that are aligned relative to a defined sequence landmark. It uses three discriminitive scoring features, motif spatial confinement (MSC), motif over-representation (MOR) and motif relative entropy (MRE). These features establish if a motif is positioned in a constrained sequence interval in positive data set and absent in negative data set.
GlobPlot is a server for exploring disorder or globularity in protein sequences.GlobPlot may be useful in domain hunting efforts. The plots indicate that instances of known domains may often contain additional N- or C-terminal segments that appear ordered.
GlobPipe is a pipeline that can be used for proteome scale analysis.
BAR+ (Bologna Annotation Resource)is a server for the annotation of protein sequences relying on a comparative large-scale genome analysis across 988 species and the entire UniProt. With BAR+ and a sequence/set of sequences (maximum number per run=50) you can annotate when possible: function (Gene Ontology), structure and ligands (Protein Data Bank), structural classification (SCOP), protein domains (Pfam). Also if your sequence falls into a cluster with a structural/some structural template/s we provide an alignment towards the template/templates based on the Cluster-HMM (HMM profile) that allows you to directly compute your 3D model. Cluster HMMs (over 10.000) are available for downloading.