WordSpy is a novel, steganalysis-based approach for genome-wide motif finding. The software views regulatory regions as a stegoscript with cis-elements embedded in ‘background’ sequences. WordSpy can discover a complete set of cis-elements and facilitate the systematic study of regulatory networks.
WHICHLOCI concerns these individual based population assignment methods but presents the method looking back on itself. Trial assignments with loci one at a time allows ranking of loci in terms of their efficiency for correct population assignment and conversely their propensity to cause false assignments. Subsequent trials with increasing numbers of loci determines what minimum number of which specific loci is required in order to attain defined power for population assignment.
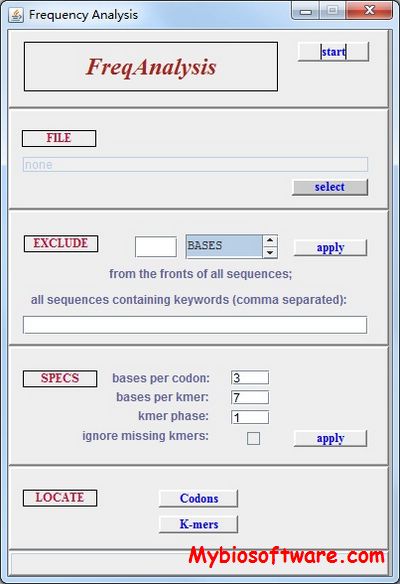
freqAnalysis was designed to identify statistically aberrant k-length nucleotide motifs in coding DNA sequences, specifically to identify putative programmed translational frameshift sites. These are short sequences capable of inducing highly efficient ribosomal frameshifting by destabilizing normal ribosome-mRNA interaction. Because of the potentially catastrophic effect of frameshifting on normal protein production, we reasoned, such sites would be selected against by evolution and hence statistically underrepresented in protein-encoding sequences.
COCO-CL (COrrelation COefficient-based CLustering) is a software for hierarchical clustering of orthology/homology relations, and identificationof orthologous groups of genes.
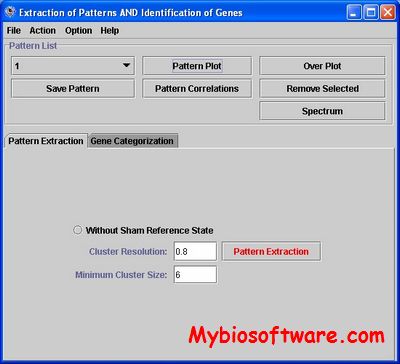
EPIG (Extracting Patterns and Identifying co-expressed Genes) is a method for Extracting microarray gene expression Patterns and Identifying co-expressed Genes. Through evaluation of the correlations among profiles, the magnitude of variation in gene expression profiles, and profile signal-to-noise ratios, EPIG extracts a set of patterns representing co-expressed genes without a pre-defined seeding of the patterns.
fdrMotif is iterative and alternates between updating the position weight matrix (PWM) and significance testing. It starts with an initial PWM and a set of sequences (e.g., from ChIP experiments). It generates many sets of background (null) sequences under the input sequence probability model. At each model estimation step, fdrMotif determines the number of binding sites in each sequence by performing statistical tests. The FDR in the original dataset is controlled by monitoring the proportion of background subsequences that are declared as binding sites. The PWM is updated using an EM algorithm with two iterative steps (the E and M steps) until convergence. In the E-step, fdrMotif normalizes the sum of the probabilities over all position s in a sequence to the number of binding sites found in the sequence.
DiNAMIC (Discovering Copy Number Aberrations Manifested In Cancer) is a novel method for assessing the statistical significance of recurrent copy number aberrations. In contrast to competing procedures, the testing procedure underlying DiNAMIC is carefully motivated, and employs a novel cyclic permutation scheme. Extensive simulation studies show that DiNAMIC controls false positive discoveries in a variety of realistic scenarios.