GeneValidator is a tool to identify problematic gene predictions based on comparisons between gene predictions and similar sequences in public databases (e.g., SwissProt).
baySeq identifies differential expression in high-throughput ‘count’ data, such as that derived from next-generation sequencing machines, calculating estimated posterior likelihoods of differential expression (or more complex hypotheses) via empirical Bayesian methods.
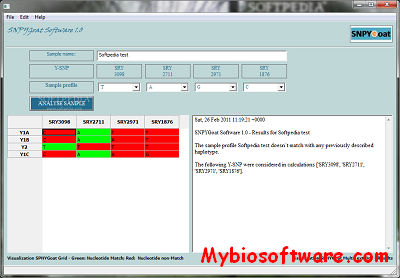
SNPYGoat Software allows users of the SNPYGoat multiplex system to rapidly identify several goat Y-chromosomal haplotypes Y1A, Y1B, Y1C and Y2 by automatically comparing the obtained profile with a reference database.
The OMiMa (the Optimized Mixture Markov model) System is a computational tool for identifying functional motifs in DNA or protein sequences. OMiMa System is based on the Optimized Mixture of Markov models that are able to incorporate most dependencies within a motif. Most important, OMiMa is capable to adjust model complexity according to motif dependency structures, so it can minimize model complexity without compromising prediction accuracy. OMiMa uses our fast Markov chain optimization method, the Directed Neighbor-Joining (DNJ), which makes OMiMa more computationally efficent.
LaneRuler will identify lanes in a gel image. The lanes on such gels may not be straight and parallel due to various reasons, and these deviations must be accounted for in order to accurately size the restriction fragments in each lane. In order to meet the high throughput requirements by the projects at the British Columbia Cancer Agency Genome Sciences Center (GSC), the software has capability to verify and correct its results automatically, and prompting for user inspection only for extremely abnormal cases. In validation testing using Bacterial Artificial Chromosome (BAC) clones, the automatic lane tracking results gave restriction fragment sizing results that are comparable to those from manually supervised lane tracking results, achieving sensitivity and specificity of restriction fragment identification exceeding 95%. The current conception of the program is able to successfully process 96% of the gels with no human intervention.
mapDamage is a computational framework written in Python and R, which tracks and quantifies DNA damage patterns among ancient DNA sequencing reads generated by Next-Generation Sequencing platforms.
TIGER is open source software for identifying rapidly evolving sites (columns in an alignment, or characters in a morphological dataset). It can deal with many kinds of data (molecular, morphological etc.). Sites like these are important to identify as they are very often removed or reweighted in order to improve phylogenetic reconstruction. When a site is changing very quickly between taxa it might not hold much phylogenetic information and therefore might simply be a source of noise. Use of TIGER can (a) allow you to see the amount of rapid evolution and noise in your alignment and (b) provide a quick and easy way to remove as many of the “noisy” sites as possible.
DIME (Differential Identification using Mixtures Ensemble) is an ensemble of methods for differential analysis. Specifically, it considers an ensemble of finite mixture models combined with a local false discovery rate (fdr) for analyzing ChIP-seq data comparing two samples. This package can also be used to identify differential in other high throughput data such as microarray and DNA methylation.