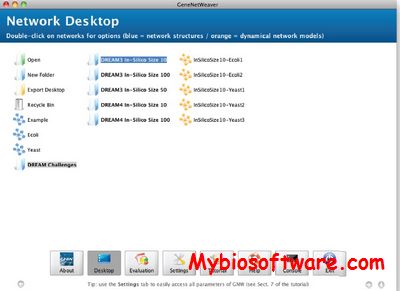
Agene automatically generates a species-specific gene predictor from a set of reliable mRNA sequences and a genome.Author applies a Hidden Markov model (HMM) that implements explicit length distribution modelling for all gene structure blocks using acyclic discrete phase type distributions. The state structure of the each HMM is generated dynamically from an array of sub-models to include only gene features represented in the training set.
CONCOORD is a method to generate protein conformations around a known structure based on geometric restrictions. Principal component analyses of Molecular Dynamics (MD) simulations of proteins have indicated that collective degrees of freedom dominate protein conformational fluctuations. These large-scale collective motions have been shown essential to protein function in a number of cases. The notion that internal constraints and other configurational barriers restrict protein dynamics to a limited number of collective degrees of freedom has led to the design of the CONCOORD method to predict these modes without doing explicit, more CPU intensive, MD simulations.
The HCE (Hierarchical Clustering Explorer) power analysis tool was designed to import any pre-existing microarray project, and interactively test the effects of user-defined definitions of α (significance), β (1-power), sample size, and effect size. The tool generates a filter for all probe sets or more focused ontology-based subsets, with or without noise filters that can be used to limit analyses of a future project to appropriately powered probe sets. We studied projects from three organisms (Arabidopsis, rat, human), and three probe set algorithms (MAS5.0, RMA, dChip PM/MM). We found large differences in power results based on probe set algorithm selection and noise filters. RMA provided exquisite sensitivity for low numbers of arrays, but this came at a cost of high false positive results (24% false positive in the human project studied). Our data suggests that a priori power calculations are important for both experimental design in hypothesis testing, and hypothesis generation, as well as for selection of optimized data analysis parameters.
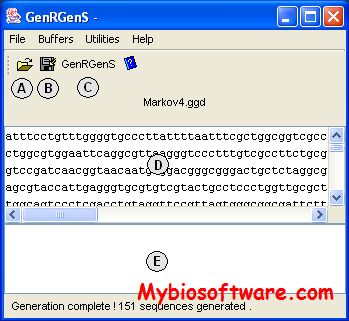
GenRGenS is a software dedicated to random generation of genomics sequences that supports several classes of models, including Markov chains, HMM, context-free grammars, PROSITE patterns and more.
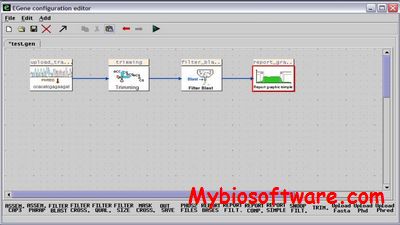
EGene is a generic, flexible and modular pipeline generation system that makes pipeline construction a modular job. EGene allows for third-party programs to be used and integrated according to the needs of distinct projects and without any previous programming or formal language experience being required.
Coed is a visual tool to facilitate pipeline construction and documentation.