CLC Genomics Workbench, for analyzing and visualizing Next Generation Sequencing data, incorporates cutting-edge technology and algorithms, while also supporting and integrating with the rest of your typical NGS workflow.
CLASS is a software tool for accurately assembling splice variants using local read coverage patterns of RNA-seq reads, contiguity constraints from read pairs and spliced reads, and optionally information about gene structure extracted from cDNA sequence databases.
SCARF is a next-gen sequence assembly tool for evolutionary genomics. Designed especially for assembling 454 EST sequences against high quality reference sequences from related species.
BugBuilder is a pipeline for the automated assembly and annotation of microbial genomes from high-throughput sequence data. It is configurable so as not to be tied to any assembler or scaffolder, and is designed to run in a cluster environment facilitating high-throughput processing of genomes.
MAXIMUS is a genome assembly pipeline which takes the best out of multiple reference assemblies and de novo assembly. The benefits of this approach include better assembled repetitive regions, less gaps and higher accuracy for the resultant assembly.
The est2assembly platform is the only platform for standardising transcriptome projects: go from raw trace files to an annotated GBrowse interface driven by the Seqfeature database. It accepts both Sanger and 454 sequencing technology for a denovo assembly, annotation and data mining of EST data.
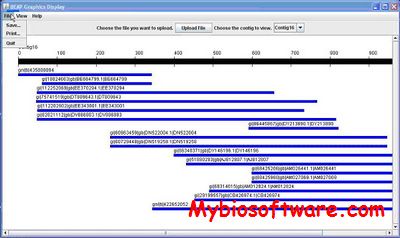
The BEAP is a computer program that uses a short starting DNA fragment, often a EST or partial gene segment, as “primer”, to recursively blast nucleotide databases in an attempt to obtain all sequences that overlaps, directly or indirectly, with the “primer” therefore help to “extend” the length of the original sequence for constructing a “full length” sequence for functional analysis, or at least to obtain neighboring regions of the segment for SNP discovery and linkage disequilibrium analysis.
BRANCH is a software that extends de novo transfrags and identifies novel transfrags with DNA contigs or genes of close related species. BRANCH discovers novel exons first and then extends/joins fragmented de novo transfrags, so that the resulted transfrags are more complete.