trace2dbEST process raw sequenceing chromatograph trace files from EST projects into quality-checked sequences, ready for submission to dbEST. trace2dbEST guides you through the creation of all the necessary files for submission of ESTs to dbEST.
::DEVELOPER
The Blaxter Lab at The Institute of Evolutionary Biology University of Edinburgh
Sybil is a web-based software package for comparative genomics, whose primary goal is to facilitate the analysis and visualization of comparative genome data, with a particular emphasis on protein and gene cluster data. Herein, a two-phase protein clustering algorithm, used to generate protein clusters suitable for analysis through Sybil and a method for creating graphical displays of protein or gene clusters that span multiple genomes are described. When combined, these two relatively simple techniques provide the user of the Sybil software (The Institute for Genomic Research [TIGR] Bioinformatics Department) with a browsable graphical display of his or her “input” genomes, showing which genes are conserved based on the parameters supplied to the protein clustering algorithm. For any given protein cluster the graphical display consists of a local alignment of the genomes in which the clustered genes are located. The genomes are arranged in a vertical stack, as in a multiple alignment, and shaded areas are used to connect genes in the same cluster, thus displaying conservation at the protein level in the context of the underlying genomic sequences. The authors have found this display-and slight variants thereof-useful for a variety of annotation and comparison tasks, ranging from identifying “missed” gene models or single-exon discrepancies between orthologous genes, to finding large or small regions of conserved gene synteny, and investigating the properties of the breakpoints between such regions.
VariantClassifier is a software tool for hierarchically classifying variants based on the genome annotation that is provided. Instead of looking at a region of the genome and seeing all the features relative to each other on the genomic axis, the VariantClassifier inverts the process so that novel variants can be tested for interest, based on the known features on the genomic axis. Furthermore, our hierarchical classification provides a prioritization of the variants that should be considered for more intensive study.
DIYA (Do-It-Yourself Annotator) is a modular and configurable open source pipeline framework, written in Perl, used for the rapid annotation of microbial genome sequences. The software is currently used to take nucleotide sequence contigs as input, either in the form of complete genomes or the result of shotgun sequencing, and produce an annotated sequence.
GeneSplicer is a fast, flexible system for detecting splice sites in the genomic DNA of various eukaryotes. The system has been trained and tested successfully on Plasmodium falciparum (malaria), Arabidopsis thaliana, human, Drosophila, and rice . Training data sets for human and Arabidopsis thaliana are included.
JIGSAW is a program designed to use the output from gene finders, splice site prediction programs and sequence alignments to predict gene models. The program provides an automated way to take advantage of the many succsessful methods for computational gene prediction and can provide substantial improvements in accuracy over an individual gene prediction program.
ExAlt is a software program designed to predict alternatively spliced overlapping exons in genomic sequence. The program works in several ways depending on the available input. ExAlt can use information about existing gene structure as well as sequence conservation to improve the precision of its predictions. ExAlt can also make predictions when only a single genomic sequence is available.
TWAIN is a software to predict genes simultaneously in two closely related eukaryotic organisms.TWAIN is a new syntenic genefinder which employs a Generalized Pair Hidden Markov Model (GPHMM) to predict genes in two closely related eukaryotic genomes simultaneously.It utilizes the MUMmer package to perform approximate alignment before applying a GPHMM based on an enhanced version of the TigrScan gene finder.
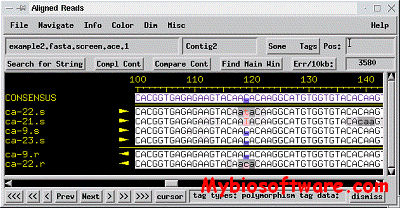
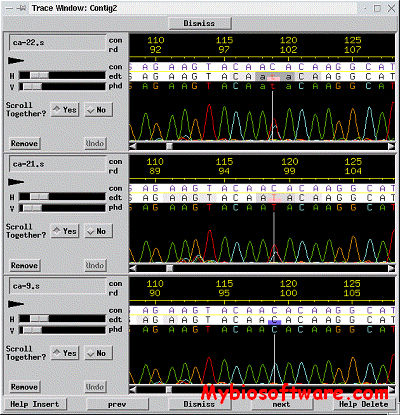
Refcomp was designed to analyze sequencing traces which contains data from strictly homozygous samples (eg. cloned DNA, mitochondrial DNA, etc.). This data represents a special case which can be analyzed for mismatches with a known reference sequence. Refcomp will determine the high quality positions within an assembled DNA contig and produce a report listing sites which differ from a defined reference sequence.
Refcomp is designed as a member of an integrated suite of sequence analysis applications which includes Phred,Phrap and Consed, and is not a stand alone program.
RefComp is available for free to researchers at academic and non-profit institutions. To aquire RefComp, please Read the Academic License Agreement, and fill in and submit the request form.
PolyPhred is a program that compares fluorescence-based sequences across traces obtained from different individuals to identify heterozygous sites for single nucleotide substitutions. PolyPhred is not a stand alone application. PolyPhred’s functions are integrated with the use of three other programs: Phred, Phrap, and Consed . PolyPhred identifies potential heterozygotes using the base calls and peak information provided by Phred and the sequence alignments provided by Phrap. Potential heterozygotes identified by PolyPhred are marked for rapid inspection using the Consed tool.