PyRx 0.9.8
:: DESCRIPTION
PyRx is a Virtual Screening software for Computational Drug Discovery that can be used to screen libraries of compounds against potential drug targets. PyRx enables Medicinal Chemists to run Virtual Screening from any platform and helps users in every step of this process – from data preparation to job submission and analysis of the results. While it is true that there is no magic button in the drug discovery process, PyRx includes docking wizard with easy-to-use user interface which makes it a valuable tool for Computer-Aided Drug Design. PyRx also includes chemical spreadsheet-like functionality and powerful visualization engine that are essential for Rational Drug Design.Visit Videos page for Getting Started Screencasts. See also Starting Virtual Screening and Getting Started with PyRx tutorials.
::DEVELOPER
Molecular Graphics Laboratory , The Scripps Research Institute
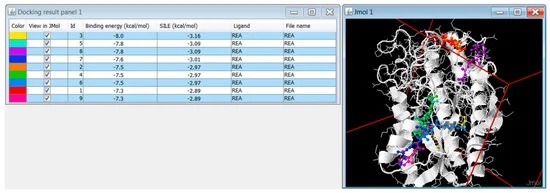
:: SCREENSHOTS

:: REQUIREMENTS
- Windows / MacOsX / Linux
:: DOWNLOAD

:: MORE INFORMATION
Citation
L.K. Wolf,
New software and Websites for the Chemical Enterprise,
Chemical & Engineering News 87, 31 (2009)