Twine 20130802
:: DESCRIPTION
Twine is a Java GUI with multiple graphical representations (‘Views’) of enhancer alignments that displays motifs, as IUPAC consensus sequences or position frequency matrices, in the context of phylogenetic conservation to facilitate cis-regulatory element discovery.
::DEVELOPER
:: SCREENSHOTS
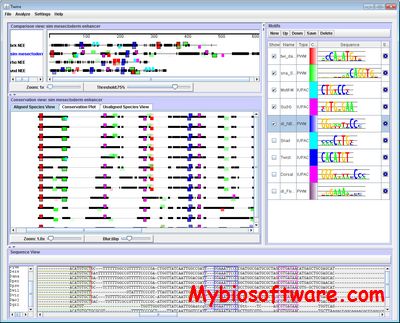
:: REQUIREMENTS
- Windows/MacOsX/Linux
- Java
:: DOWNLOAD

:: MORE INFORMATION
Citation
Bioinformatics. 2013 Jul 1;29(13):1690-2. doi: 10.1093/bioinformatics/btt264. Epub 2013 May 8.
Twine: display and analysis of cis-regulatory modules.
Pearson JC, Crews ST.