DiagAF is a more accurate and efficient pre-alignment filter for sequence alignment. It can efficiently filter out candidates that contain errors greater than the edit distance threshold during read mapping.
MUMmerGPU is an open-source high-throughput parallel pairwise local sequence alignment program that runs on commodity Graphics Processing Units (GPUs) in common workstations. MUMmerGPU uses the new Compute Unified Device Architecture (CUDA) from nVidia to align multiple query sequences against a single reference sequence stored as a suffix tree. By processing the queries in parallel on the highly parallel graphics card, MUMmerGPU achieves more than a 10-fold speedup over a serial CPU version of the sequence alignment kernel, and outperforms the exact alignment component of MUMmer on a high end CPU by 3.5-fold in total application time when aligning reads from recent sequencing projects using Solexa/Illumina, 454, and Sanger sequencing technologies. MUMmerGPU is a low cost, ultra-fast sequence alignment program designed to handle the increasing volume of data produced by new, high-throughput sequencing technologies.
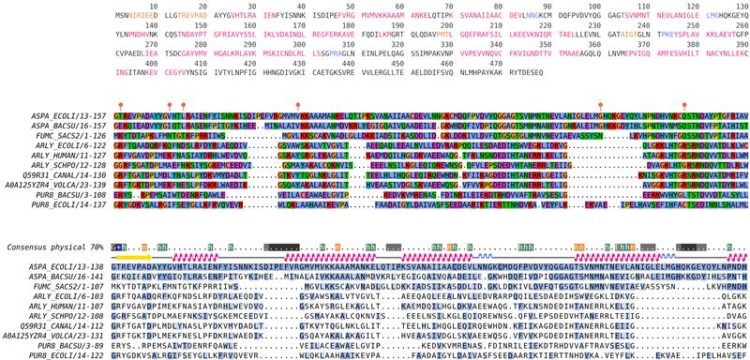
SIBIS (Bayesian Inconsistency in Sequences) is designed to detect such inconsistencies based on the evolutionary information in multiple sequence alignments. A Bayesian framework, combined with Dirichlet mixture models, is used to estimate the probability of observing specific amino acids and to detect inconsistent or erroneous sequence segments.
FSTVAL is an open access web tool to manage bulk flanking sequence tags (FSTs).FSTVAL automatically evaluates the FSTs and finds the best mapping positions of the FST against a known genome sequence.
rvsel is an R package for rare variants selection with sequence data. The most outome-related rare variants are selected within a gene or a genetic region. The selection procedure is based on the power set of the subset of the rare variants.