Gbench (NCBI Genome Workbench) is an integrated application for viewing and analyzing sequence data. With Genome Workbench, you can view data in publically available sequence databases at NCBI, and mix this data with your own private data.Genome Workbench can display sequence data in many ways, including graphical sequence views, various alignment views, phylogenetic tree views, and tabular views of data. It can also align your private data to data in public databases, display your data in the context of public data, and retrieve BLAST results.
ASAP (Advanced Sequence Automated Pipeline)is an open source pipeline designed to assist users in managing various jobs associated with processing HiSeq data on a cluster or in serial. . The software was designed to alleviate these issues by providing a modular system to allow users with different needs to process their data with a minimal amount of effort. In addition to minimizing human involvement, ASAP is designed to work on the researcher’s local computer cluster, if one is available
SEQMINER is an R package for annotating and querying files of sequence variants (e.g., VCF/BCF files) and summary association statistics (e.g., METAL/RAREMETAL files), and for integrating bioinformatics databases.
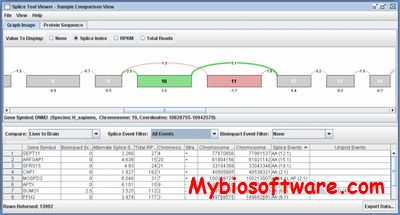
SpliceSeq provides a quick, easy method of investigating alternative mRNA splicing in next generation mRNA sequence data. The tool may be used on a single mRNA-Seq sample to identify genes with multiple spliceforms or on a pair of samples to identify differential splicing between the samples. Sequence reads are mapped to splice graphs that unambiguously quantify the inclusion level of each exon and splice junction. The graphs are then traversed to predict the protein isoforms that are likely to result from the observed exon and splice junction reads. UniProt annotations are mapped to each protein isoform to identify potential functional impacts of alternative splicing.
ClonalFrame is a computer package for the inference of bacterial microevolution using multilocus sequence data.In a nutshell, ClonalFrame identifies the clonal relationships between the members of a sample, while also estimating the chromosomal position of homologous recombination events that have disrupted the clonal inheritance.ClonalFrame can be applied to any kind of sequence data, from a single fragment of DNA to whole genomes. It is well suited for the analysis of MLST data, where 7 gene fragments have been sequenced, but becomes progressively more powerful as the sequenced regions increase in length and number up to whole genomes. However, it requires the sequences to be aligned. If you have genomic data that is not aligned, we recommend using Mauve which produces alignment of whole bacterial genomes in exactly the format required for analysis with ClonalFrame.
ClonalFrameML is a software package that performs efficient inference of recombination in bacterial genomes.
SeqToolBox is a set of Perl modules and scripts for day to day operations on sequence data. Includes, parsers for standard file formats and some utility modules, such as modules for working with “sets”, a set of modules for handling NCBI taxonomy database etc. The bin directory contains some scripts to run jobs on clusters.