ScripTree 17
:: DESCRIPTION
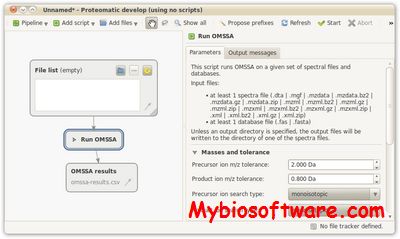
ScripTree would enable the saving of graphical analyses involving numerous and complex tree handling operations and would allow the automation of repetitive tasks.ScripTree is a tool intended to fill this gap. It is an interpreter to be used in batch mode. Phylogenetic graphics instructions are stored in a text file and performed in a sequential way. Such instructions are related to tree rendering as well as tree annotation.
::DEVELOPER
:: SCREENSHOTS
N/A
:: REQUIREMENTS
- Linux / Mac OsX / Windows
:: DOWNLOAD

:: MORE INFORMATION
Citation
Bioinformatics. 2010 Apr 15;26(8):1125-6. Epub 2010 Mar 1.
ScripTree: scripting phylogenetic graphics.
Chevenet F, Croce O, Hebrard M, Christen R, Berry V.