DupHMM
:: DESCRIPTION
DupHMM is an easy to use copy number variant identifier
::DEVELOPER
Korf Lab at the University of California, Davis.
:: SCREENSHOTS
N/A
:: REQUIREMENTS
- Windows/MacOsX/Linux
- Python
- R package
:: DOWNLOAD

:: MORE INFORMATION
:: DESCRIPTION
Picard comprises Java-based command-line utilities that manipulate SAM files, and a Java API (SAM-JDK) for creating new programs that read and write SAM files. Both SAM text format and SAM binary (BAM) format are supported.
:: DEVELOPER
:: SCREENSHOTS
N/A
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
:: DESCRIPTION
AgileSAMFileSorter reads the sequence reads in an unsorted *.sam file and creates a second *.sam file in which all the sequence reads are sorted by chromosome number and position.
::DEVELOPER
:: SCREENSHOTS
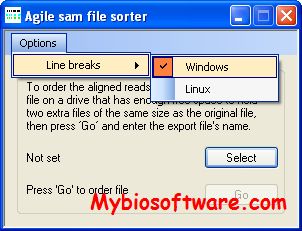
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION
:: DESCRIPTION
AgileAnnotator identifies sequence variants that repeatedly occur in reads that have previously been aligned to a reference sequence by an alignment program, such as BWA or Novoalign, and exported as SAM files.
::DEVELOPER
:: SCREENSHOTS
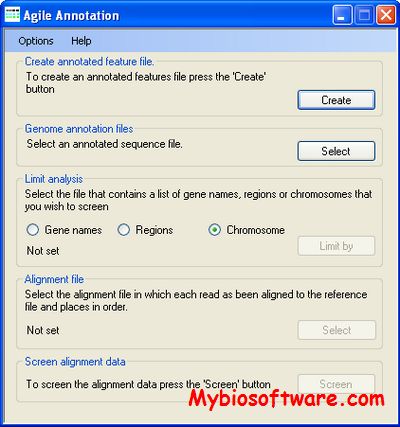
:: REQUIREMENTS
:: DOWNLOAD
:: MORE INFORMATION