pFind Studio 2.8.8
:: DESCRIPTION
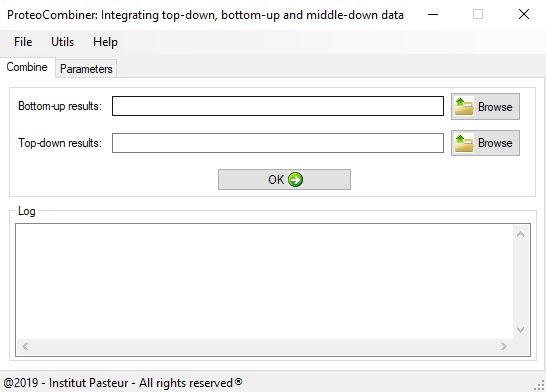
pFind Studio is a computational solution for mass spectrometry-based proteomics. pFind Studio includes pFind, pBuild, pLabel, pXtract, pParse and pScan
pXtract creates .DTA .MGF and .MS2 input files directly from Thermo Scientific .raw LC-MS/MS data files.
pParse is a software dedicated to recalibrate the monoisotopic of precursors in MS/MS spectra datasets assigned by mass spectrometry
pFind is a search engine for peptide and protein identification via tandem mass spectrometry.
pNovo+ is a de novo peptide sequencing algorithm using complementary HCD and ETD tandem mass spectra.
pBuild is a tool that can compare several search engines’ results and combine them together. The latest version, pBuild v2.0, can process the search results of pFind, SEQUEST and Mascot.
pQuant is the software for quantitative proteomics, evaluates the accuracy of caluculated peptide and protein ratios.
pLabel is a spectra labeling tool that can visualize the global- and local-view peptide-spectrum matches, given the results of pFind or any other search engines. pLabel can label both CID and ETD spectra, and implement the manual de novo sequencing.
pLink is a software dedicated for the analysis of chemically cross-linked proteins or protein complexes using mass spectrometry.
pScan is a flexible tool that helps biologists to preprocess protein sequence databases in proteomics research.
pCluster is a software tool aiming at detecting protein modifications independent of sequence databases by tandem mass spectral clustering.
pMatch is a spectral library search tool, which is deliberately designed for the open search mode.
::DEVELOPER
Bioinformatics group, Institute of Computing Technology, Chinese Academy of Sciences
:: SCREENSHOTS

:: REQUIREMENTS
:: DOWNLOAD
 pFind Studio
pFind Studio
:: MORE INFORMATION
Citation
Rapid Communications in Mass Spectrometry, 21,2985-2991,2007.
pFind 2.0: a software package for peptide and protein identification via tandem mass spectrometry.
Le-Heng Wang, De-Quan Li, Yan Fu, Hai-Peng Wang, Jing-Fen Zhang, Zuo-Fei Yuan,Rui-Xiang Sun, Rong Zeng, Si-Min He, Wen Gao.