NJplot 2.4
:: DESCRIPTION
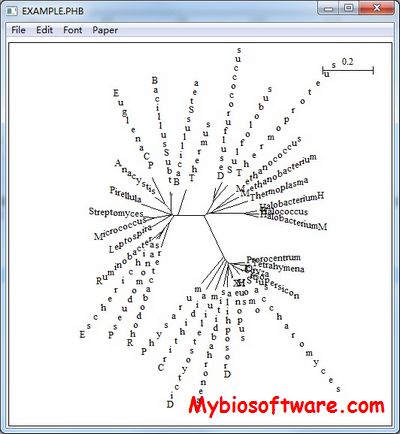
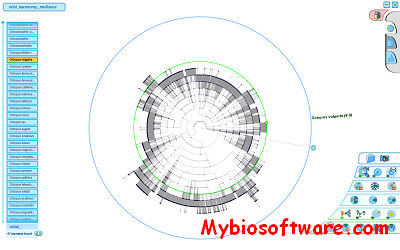
NJplot is a tree drawing program able to draw any phylogenetic tree expressed in the Newick phylogenetic tree format (e.g., the format used by the PHYLIP package). NJplot is especially convenient for rooting the unrooted trees obtained from parsimony, distance or maximum likelihood tree-building methods.
::DEVELOPER
:: SCREENSHOTS

:: REQUIREMENTS
- Windows / Linux / MacOSX
:: DOWNLOAD

:: MORE INFORMATION
Citation
Perrière, G. and Gouy, M. (1996)
WWW-Query: An on-line retrieval system for biological sequence banks.
Biochimie, 78, 364-369.