Raster3D 3.0-7
:: DESCRIPTION
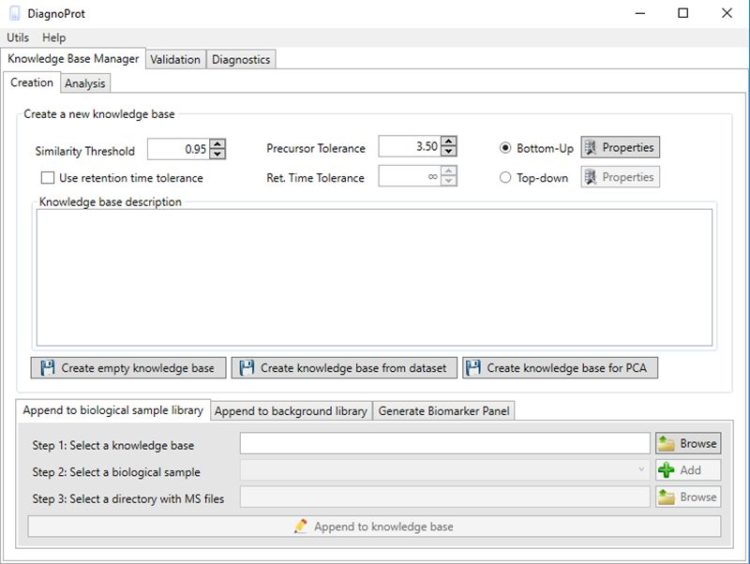
Raster3D is a set of tools for generating high quality raster images of proteins or other molecules. The core program renders spheres, triangles, cylinders, and quadric surfaces with specular highlighting, Phong shading, and shadowing. It uses an efficient software Z-buffer algorithm which is independent of any graphics hardware. Ancillary programs process atomic coordinates from PDB files into rendering descriptions for pictures composed of ribbons, space-filling atoms, bonds, ball+stick, etc. Raster3D can also be used to render pictures composed in other programs such as Molscript in glorious 3D with highlights, shadowing, etc. Output is to pixel image files with 24 bits of color information per pixel.
::DEVELOPER
:: SCREENSHOTS
N/A
:: REQUIREMENTS
- MacOsx/Linux/Windows
:: DOWNLOAD

:: MORE INFORMATION
Citation
Merritt, Ethan A. and Bacon, David J. (1997).
“Raster3D: Photorealistic Molecular Graphics”
Methods in Enzymology 277, 505-524.