MDI-GPU is an improved implementation of a Bayesian correlated clustering algorithm, that permits integrated clustering to be routinely performed across multiple datasets, each with tens of thousands of items.
Memoir (MEMbrane prOteIn modelleR) is a homology modelling algorithm designed for membrane proteins. The inputs are the sequence which is to be modelled, and the 3D structure of a template membrane protein.
SRSim is constructed from the molecular dynamics simulator LAMMPS and a set of extensions for modeling rule-based reaction systems. The aim of this software is coping with reaction networks that are combinatorially complex as well as spatially inhomogeneous. On the one hand, there is a combinatorial explosion of necessary species and reactions that occurs when complex biomolecules are allowed to interact, e.g. by polymerization or phosphorilation processes. On the other hand, diusion over longer distances in the cell as well as the geometric structures of sophisticated macromolecules can further in uence the dynamic behavior of a system. Addressing the mentioned demands, the SRSim simulation system features a stochastic, particle based, spatial simulation of Brownian Dynamics in three dimensions of a rule-based reaction system.
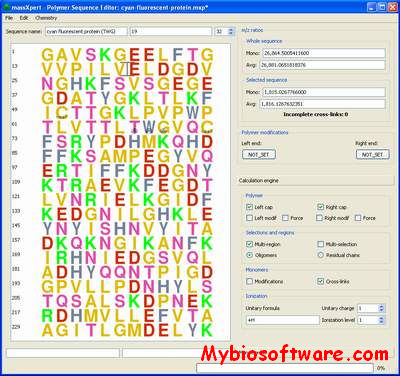
massXpert is a cross-platform software environment for polymer chemistry modelling and simulation/analysis of mass spectrometric data.The massXpert project aims at providing (bio)chemists with a software package allowing the following:
User-specific atom definitions and polymer chemistry definitions;
Powerful sequence editing with user-definied glyphs for each monomer and monomer chemical modification;
Polymer sequence chemical/enzymatic cleavage;
Gas-phase fragmentation of oligomers;
Mass-to-charge ratio calculations with inline change of ionization agent.
Bio-PEPA is a language for the modelling and the analysis of biochemical networks.It is based on PEPA, a process algebra originally defined for the performance analysis of computer systems, and extends it in order to handle some features of biochemical networks, such as stoichiometry and different kinds of kinetic laws. A main feature of Bio-PEPA is the possibility to support different kinds of analysis, including stochastic simulation, analysis based on ordinary differential equations (ODEs) and model checking in PRISM.
Bio-PEPA Workbench is an implementation of Bio-PEPA which allows modellers to write models in the Bio-PEPA language and to animate them using stochastic simulation.
BCT is a text-based, deterministic simulator of the chemotaxis signal transduction pathway in E. coli. The BCT (bacterial chemotaxis) model is built from units representing the molecular components in the pathway, almost all of which can be assigned experimentally determined intracellular concentrations and enzymatic rate or binding constants.