PSAAM
:: DESCRIPTION
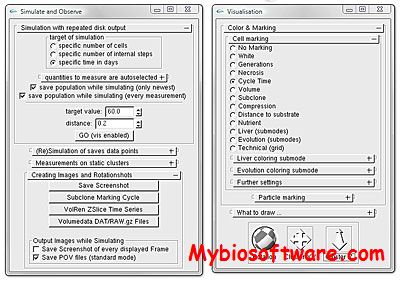
PSAAM (Protein Sequence Analysis And Modelling) includes many useful features for analysis of structural information from sequence data, cartoons for graphical representation of secondary structures, prediction programs, and prediction aids, a ribosome function for generation of coordinates sets in PDB format (including Header information) for viewing by molecular graphics packages (examples, see also PDVWIN), and a SeqPlot function for plotting secondary structural models (helical cylinders and wheels, coils, etc., with each residue indicated by a circle, color-coded (or coded by line thickness) according to the current physico-chemical index.
::DEVELOPER
:: SCREENSHOTS

:: REQUIREMENTS
- Windows
:: DOWNLOAD

:: MORE INFORMATION