InferRho is a MCMC based program for jointly estimating the crossing-over and gene-conversion parameters from population genomic datasets under a Bayesian framework.
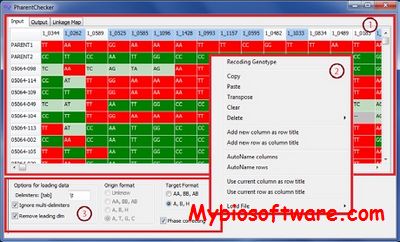
ParentChecker is a user-friendly tool that uses the segregation patterns of progeny to infer missing genotype information of parent lines that have been used to construct mapping populations. It can also be used to automate correction of linkage phase errors in genotypic data that is in ABH format.
Ancestor is a software which infer ancestral amino acid sequences from a set of homologous amino acid sequences whose phylogenetic relationships are known.
Anc-gene is a software for inference of ancestral nucleotide sequences of protein coding genes from a set of present-day sequences whose phylogenetic relationships are known. It first infers the amino acids by the distance-based Bayesian method, and then infers the underlying nucleotide sequences by fixing the inferred amino acids.
HMC (Haplotype Inference Based on Markov Chain)is a new haplotype inference methods based on Markov chain model which do not assume haplotype blocks in the population and allows each individual haplotype to have its own structure, thus are able to accommodate recombination and obtain higher adaptivity to the genotype data, specifically in the case of long marker maps. The proposed method presents a general Markov chain model for haplotype inference problem. A dynamic programming algorithm is developed for the model. The algorithm is theoretically guaranteed to find exact global optimal solutions within polynomial running time. Through extensive computational experiments on simulated and real genotype data, the designed algorithm is shown to be efficient, and outperforms previous methods.
Ling-Yun Wu, Ji-Hong Zhang, and Raymond Chan. Improved approach for haplotype inference based on Markov chain.
In Proceedings of 2nd International Symposium on Optimization and Systems Biology, Lecture Notes in Operations Research, Vol. 9, pp. 204–215, World Publishing Corporation, Beijing, 2008.
NET-SYNTHESIS performs combined synthesis, inference and simplification of signal transduction networks. The main idea of the application lies in representing observed indirect causal relationships as network paths, introducing pseudo-vertices for unknown intermediaries of these paths and using techniques from combinatorial optimization to find the most parsimonious graph consistent with all experimental observations. The software contains algorithms for (i) transitive reduction of an initially synthesized graph subject to the constraints that no edges corresponding to direct interactions are eliminated and (ii) pseudo-vertex collapse subject to the constraints that real (known) vertices are not eliminated.
IPHULA / pIPHULA (parallel Inference of Population History Using a Likelihood Approach) is a software that investigates parameters of population history based on a Monte Carlo simulation using the maximum likelihood framework.
SNAP (Suite of Nucleotide Analysis Programs) Workbench is a Java program that manages and coordinates a series of analysis programs for making inferences on population processes. SNAP workbench allows the user to customize the implementation of complex console programs and functions for the purpose of automating and enhancing data exploration. In our implementation, the workbench facilitates population parameter estimation by ensuring that the assumptions and program limitations of each analysis method are met and by providing a step-by-step methodology to effectively integrate both summary-statistic methods and coalescent-based population genetic models.
SNAP Map is a command line-based tool that collapses DNA sequence data into unique haplotypes, extracts variable sites, and manipulates output into multiple formats for input into existing software packages for evolutionary analyses. Map includes novel features such as recoding indels, including or excluding variable sites that violate an infinite-sites model and the option of collapsing sequences with corresponding phenotypic information, important in testing for significant haplotype-phenotype associations.
SNAP Combine is a command-line based tool that merges the contents of multiple single locus DNA sequence files into a single multi-locus output file. There are various input and output file formats. The files can be merged into a union or intersection of all the input loci. Additionally Combine tracks the start and end positions of each file allowing the user to exclude variable sites or taxa, important in creating input files for multilocus analyses.