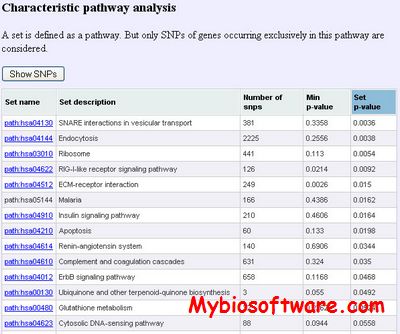
GWAS Pathway Identifier combines GWAS(Genome-Wide Accociation Studies) and pathway data as well as known and predicted protein-interaction data to identify disease specific SNP sets.
Refge is a Global Alliance for Genomics and Health API specification to access reference sequences and sub-sequences using an identifier derived from the sequence itself.
Kinannote identifies and classifies protein kinases in a user-provided fasta file using an HMM derived from serine/threonine protein kinases, a position specific scoring matrix derived from the HMM, and comparison with a local version of the curated kinase database from kinase.com. If the user inputs a complete proteome, additional modules evaluate the completeness of the kinome and place it in context with reference kinomes.
HaploBlock is a software program which provides an integrated approach to haplotype block identification, haplotyping SNPs (or haplotype phasing, resolution or reconstruction) and linkage disequilibrium (LD) mapping (or genetic association studies). HaploBlock is suitable for high density haplotype or genotype SNP marker data and is based on a statistical model which takes account of recombination hotspots, bottlenecks, genetic drift and mutations and has a Markov Chain at its core.
PIUS is an open source tool that performs peptide identification from tandem mass spectrograms using the six-frame translation of the complete genome. Differently from peptidomics methods that also allow a search against the complete genome, PIUS does not limit analysis of the genome to a small set of sequences that match a list of de novo reconstructions. Instead, it performs an exhaustive scan of the translation of the six reading frames of the complete genome. Therefore, this search is not biased towards a subset of candidates.
Bioinformatics. 2013 Aug 1;29(15):1913-4. doi: 10.1093/bioinformatics/btt298. Epub 2013 May 24. PIUS: peptide identification by unbiased search.
Costa EP1, Menschaert G, Luyten W, De Grave K, Ramon J.
AUDACITY is novel computational approach for the identification of Runs of Homozygosity by using VCF files from whole-exome and whole-genome sequencing data generated by second generation sequencing technologies.