PHIRE 1.00
:: DESCRIPTION
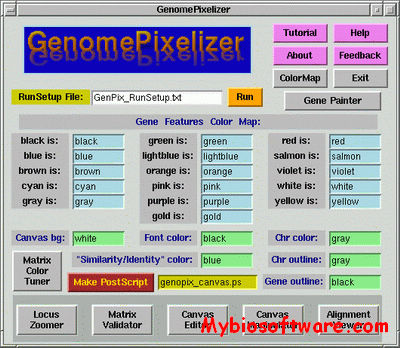
PHIRE (PHage In silico Regulatory Elements) is a standalone program in Visual Basic, performs an algorithmic string-based search on bacteriophage genome sequences, discovering and extracting blocks displaying sequence similarity, corresponding to conserved regulatory elements contained within these genomes in a systematic manner, without any prior experimental or predictive knowledge.
::DEVELOPER
:: SCREENSHOTS

:: REQUIREMENTS
- Windows
:: DOWNLOAD

:: MORE INFORMATION
Citation
Lavigne, R., Sun, W.D. and Volckaert, G.
PHIRE, a deterministic approach to reveal regulatory elements in bacteriophage genomes.
Bioinformatics (2004) 20: 629-635.