GPASS (Genome-wide Poisson Approximation for Statistical Significance) detects SNP disease associations in case control studies, controls FWER and FDR adjusting for dependence/linkage disequilibrium.
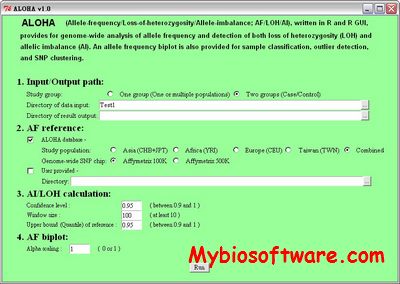
ALOHA (Allele-Frequency/Loss-of-Heterozygosity/Allelic-Imbalance) is a tool for a genome-wide analysis of allele frequency, detection of loss of heterozygosity and identification of allelic imbalance. Moreover, chromosome-wise allele frequency biplots are provided for sample classification, outlier detection and SNP clustering.
HPeak is a hidden Markov model-based approach that can accurately pinpoint regions to where significantly more sequence reads map. Testing on real data shows that these regions are indeed highly enriched by the right protein binding sites.ChIP-Seq is an important application of the massively parallel sequencing technologies aiming to identify all the locations in the genome where a specific protein binds. While direct counting of the sequencing reads can reveal many such binding sites, it is desirable to develop a statistical sound method to explicitly model the uncertainties involved for better and more interpretable results.