DisLocate is a a two-step method based on machine learning models for predicting both the bonding state and the connectivity patterns of cysteine residues in a protein chain.
CleaveLand is a command-line executed pipeline for finding cleaved small RNA targets using degradome data (also known as PARE [parallel analysis of RNA ends] and GMUCT [genome-wide mapping of uncapped transcripts]). Provided with a set of degradome data, a list of small RNA queries, a reference transcriptome/mRNA set, CleaveLand outputs potentially cleaved small RNA targets along with other supporting information.
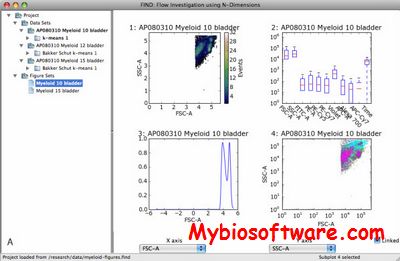
FIND is a software package designed to facilitate the visualization and analysis of Flow Cytometry data for the scientific target audience, as well as providing a target platform for developers to implement their FC-specific algorithms.
SnowShoes-FTD is a bioinformatics tool to identify fusion transcripts from paired-end transcriptome sequencing data. The tool employs multiple steps of false positive filtering and nominates the fusion candidates with high confidence (approaching 100% true positive rate). The unique features of SnowShoes-FTD include: (i) the ability to discover multiple fusion isoforms in which the two gene partners give rise to transcripts with different junctions; (ii) prediction of potential fusion mechanisms including inversion, translocation, and/or interstitial deletions; (iii) identification of whether the junction point in a fusion transcript occurs at the boundaries of known exons which implies the fusion events might have happened inside an intron in DNA and transcribed to the fusion transcript.
::DEVELOPER
Bioinformatics Program, Division of Biomedical Statistics and Informatics, Mayo Clinic Research
:: SCREENSHOTS
N/A
:: REQUIREMENTS
Linux
Perl
:: DOWNLOAD
Please contact the author, Yan W. Asmann, Ph.D. smann.yan@mayo.edu
FOOTER analyses a pair of homologous mammalian DNA sequences (i.e. human and mouse/rat) for high probability binding sites of known transcription factors. A set of Position-Specific Scoring Matrices (PSSM) has been carefully constructed from mammalian transcription factor binding sites deposited in TRANSFAC database.
LBL (Logistic Bayesian Lasso) is an R package that performs Logistic Bayesian Lasso for finding association of SNP haplotypes with a trait in a case-control setting. Bayesian lasso is used to find the posterior distributions of logistic regression coefficients, which are then used to calculate Bayes Factor to test for association with haplotypes.
LBLGXE expands on the original LBL. Gene-environment interaction (GXE)