Tracembler
:: DESCRIPTION
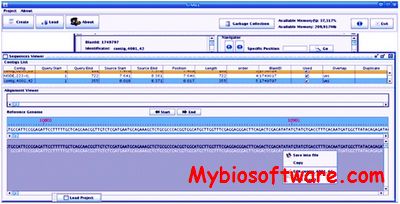
Tracembler takes one or multiple DNA or protein sequence(s) as input to the NCBI Trace Archive BLAST engine to identify matching sequence reads from a species of interest. The BLAST searches are carried out recursively such that BLAST matching sequences identified in previous rounds of searches are used as new queries in subsequent rounds of BLAST searches. Tracembler streamlines the process of recursive database searches, sequence assembly, and gene identification in resulting contigs in attempts to identify homologous loci of genes of interest in species with emerging whole genome shotgun reads.
::DEVELOPER
The Brendel Group @ Indiana University
:: SCREENSHOTS
N/A
:: REQUIREMENTS
- Linux
:: DOWNLOAD

:: MORE INFORMATION
Citation
Dong, Q., Wilkerson, M.D. & Brendel, V. (2007)
Tracembler – software for in silico chromosome walking in unassembled genomes.
BMC Bioinformatics 8, 151.