DiagAF is a more accurate and efficient pre-alignment filter for sequence alignment. It can efficiently filter out candidates that contain errors greater than the edit distance threshold during read mapping.
Harvest is a suite of core-genome alignment and visualization tools for quickly analyzing thousands of intraspecific microbial genomes. Harvest includes: Parsnp, a fast core-genome multi-aligner, Gingr, a dynamic visual platform, and harvest-tools, providing both a reference compressed binary archive and format conversion tools.
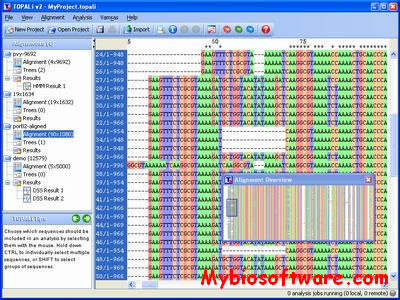
Topali (tree TOPology-related analysis of ALignments Interface) is a software for statistical and evolutionary analysis of multiple sequence alignments.The extended TOPALi v2 provides phylogenetic model selection, Bayesian analysis (BA) and Maximum Likelihood (ML) phylogenetic tree estimation, detection of sites under positive selection, and recombination breakpoint location analysis.
MUMmerGPU is an open-source high-throughput parallel pairwise local sequence alignment program that runs on commodity Graphics Processing Units (GPUs) in common workstations. MUMmerGPU uses the new Compute Unified Device Architecture (CUDA) from nVidia to align multiple query sequences against a single reference sequence stored as a suffix tree. By processing the queries in parallel on the highly parallel graphics card, MUMmerGPU achieves more than a 10-fold speedup over a serial CPU version of the sequence alignment kernel, and outperforms the exact alignment component of MUMmer on a high end CPU by 3.5-fold in total application time when aligning reads from recent sequencing projects using Solexa/Illumina, 454, and Sanger sequencing technologies. MUMmerGPU is a low cost, ultra-fast sequence alignment program designed to handle the increasing volume of data produced by new, high-throughput sequencing technologies.
PAcAlCI is novel intelligent algorithm based on least square support vector machine (LS-SVM) to predict how accurately ten different MSA tools could align a particular set of sequences.
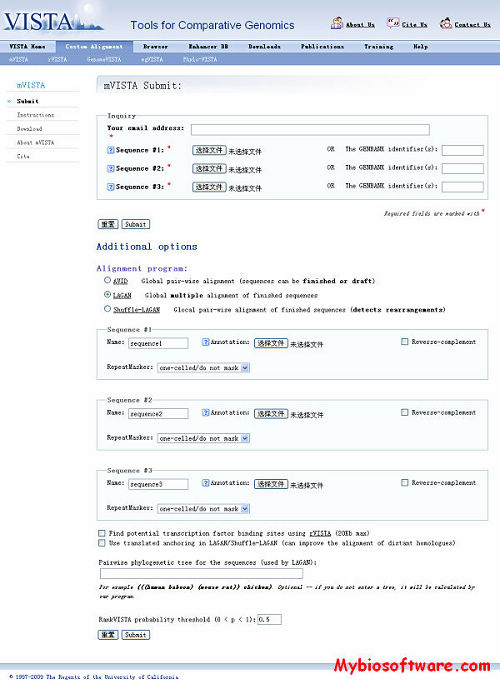
mVISTA is a set of programs for comparing DNA sequences from two or more species up to megabases long and visualize these alignments with annotation information. mVISTA has a clean output, allowing for easy identification of sequence similarities and differences, and is easily configurable, enabling the visualization of alignments of various lengths at different levels of resolution. It is implemented as an on-line server that provides access to global pairwise, multiple and glocal (global with rearrangements) alignment tools.
mVISTA and Avid are free for academic or non-profit research institutions to use for internal research purposes. Commercial companies may submit sequences over the Web using the VISTA web site at no charge. To obtain a copy of mVISTA or Avid for internal use, commercial entities are required to purchase a site license for commercial use.
Citation:
Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 2004 Jul 1;32(Web Server issue):W273-9
Mayor C., Brudno M., Schwartz J. R., Poliakov A., Rubin E. M., Frazer K. A., Pachter L. S. and Dubchak I. (2000) VISTA: Visualizing Global DNA Sequence Alignments of Arbitrary Length. Bioinformatics, 16:1046.