HECTOR 1.0
:: DESCRIPTION
HECTOR is a parallel multistage homopolymer spectrum based error corrector for 454 sequencing data. In this algorithm, for the first time we have investigated a novel homopolymer spectrum based approach to handle homopolymer insertions or deletions, which are the dominant sequencing errors in 454 short-reads.
::DEVELOPER
:: SCREENSHOTS
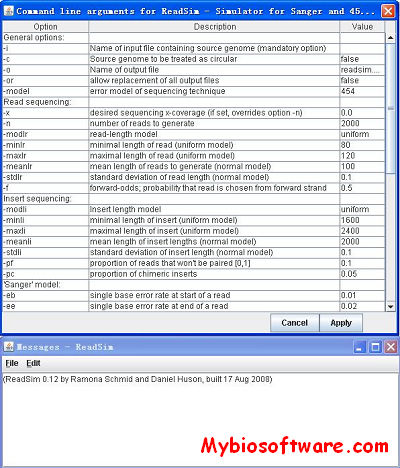
N/A
:: REQUIREMENTS
- Linux
:: DOWNLOAD

:: MORE INFORMATION
Citation
BMC Bioinformatics. 2014 May 6;15(1):131. [Epub ahead of print]
HECTOR: a parallel multistage homopolymer spectrum based error corrector for 454 sequencing data.
Wirawan A, Harris RS, Liu Y, Schmidt B, Schröder J.