jagn 1.02
:: DESCRIPTION
jagn is a software for an Artificial Gene Networks (AGNs) model generation through theoretical models of complex networks, which is used to simulate temporal expression data, which can be used by computational methods to recover the network topology, and then, analyse the results based on complex networks measurements/topology.
::DEVELOPER
:: SCREENSHOTS
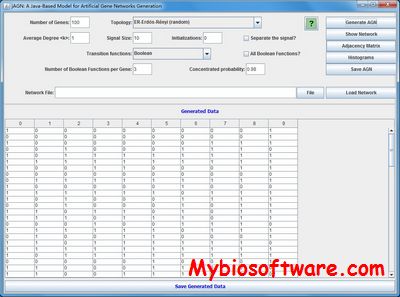
:: REQUIREMENTS
- Linux / Windows / MacOsX
- Java
:: DOWNLOAD

:: MORE INFORMATION
Citation:
Lopes, Fabrício M.; Cesar-Jr, Roberto M.; Costa, Luciano da F.
Gene expression complex networks: synthesis, identification and analysis.
Journal of Computational Biology, v. 18, p. 1353-1367, 2011.